Projects
TREP - Transposable Element Platform
The TREP database is a highly curated collection of DNA repeat sequences. It originally started as a collection of repeats from Triticeae (wheat, barley and their relatives), which is why the acronym initially stood for “Triticeae REPeats”. Over the years, sequences from animals, fungi, bacteria and viruses were included. Currently, TREP does not contain and sequences from Archeae.
The TREP database is under revision at the moment and can not be reached. Sorry for the inconveniences.
TREP contains two divisions, the database for genomic sequences (TREP) and the database for hypothetical TE proteins PTREP. The hypothetical proteins are predicted based on consensus sequences of respective TE families, with exon/intron structures being predicted based on homology to previously described TE proteins and transcriptome data (if available).
TREP contains the following types of repeats:
Transposable elements (TEs)
Most repeats deposited at TREP are TE sequences. We use the classification system proposed by Wicker et al., 2007, where TEs are classified into Classes, Orders and Superfamilies, families and subfamilies. Each superfamily is assigned a three letter code which precedes the family name. Optional subfamily designations can be added to the name. For example RLC_Angela_A is a Copia retrotransposon of the family Angela, subfamily A.
Tandem repeats
Most genomes contain short tandem repeats, which are often also called “satellite” repeats. These are short (typically 30-500 bp) units that are repeated locally many times, leading to large arrays of sequences that are repeats in the same direction (“direct” repeats). Many organisms contain tandem repeats in centromeres or near telomeres.
TREP does not contain sort sequence repeats (i.e. SSRs or microsatellites), which are simple repeats of a few bp in length (usually 1-10 bp).
Ribosomal DNA (rDNA)
In Eucaryotes, rDNA are ususally found in a single locus in the genome where the rDNA operon is repeated in tandem (direct) hundreds or thousands of times. Due to the high copy numbers, rDNA regularly pop up in repeat analyses, which is why it is useful to identify them early on.
Additionally, rDNAs are useful for taxonomic classification. The 16S rDNA gene is typically used to classify bacterial, while the highly variable internal transcribed spacers (ITS) are used for classification of eukaryotes.
tDNA
TREP also contains genes for transfer RNA (tDNAs) from a range of species.
TREP trivia
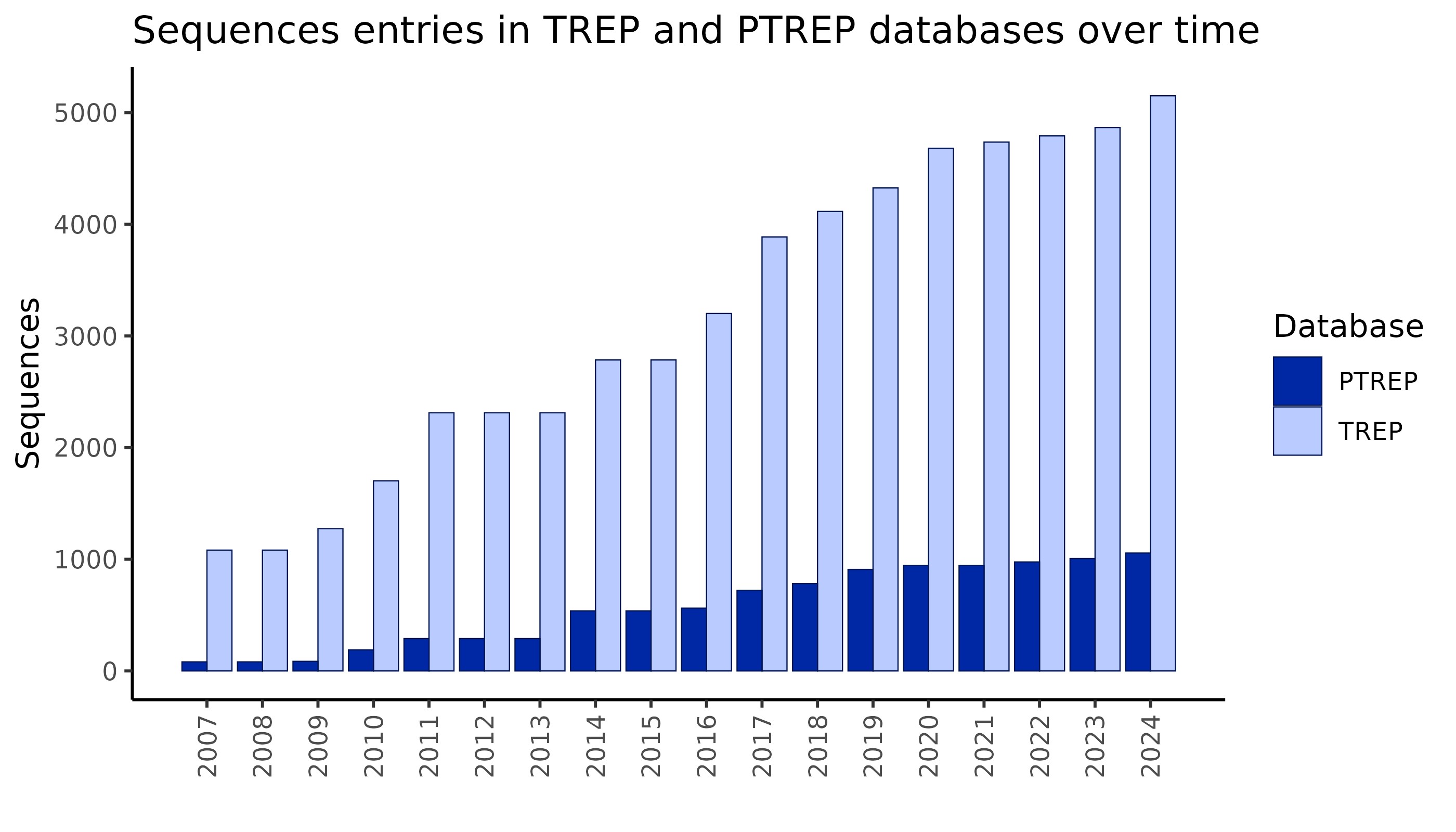
- TREP currently contains over 5,000 nucleotide sequences, representing over 1,200 repeat families.
- PTREP contains over 1,000 protein sequences, representing over 450 TE families.
- TREP contains sequences from 134 genera and 260 species.
References:
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH (2007) A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 8:973-82.
DOI: 10.1038/nrg2165
Please cite use of resources on this website as follows:
Schlagenhauf, Edith and Wicker, Thomas (2016). The TREP platform: A curated database of transposable elements. Available at https://trep-db.uzh.ch.
Plant/pathogen interactions
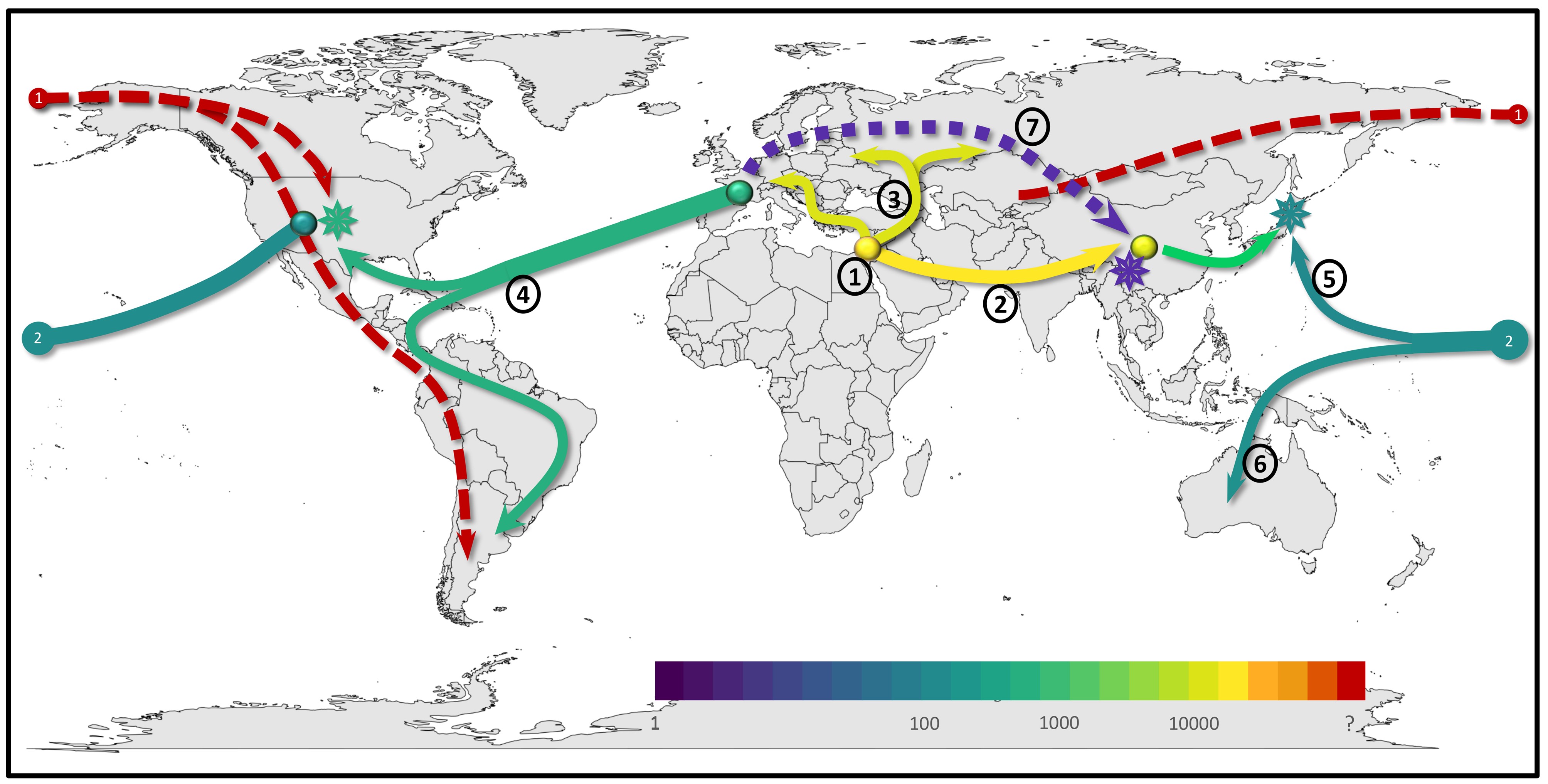
In my group, we study genomics of interactions between crop plants and their fungal pathogens. One of our model systems is wheat powdery mildew (Blumeria graminis). We have sequenced and analysed the genomes of several mildews strains. Here, we showed that the dynamic evolution of effector gene clusters is crucial for the adaptation of the pathogen to race-specific plant resistance genes. Additionally, we studied mildew that can infect “Triticale”, a synthetic hybrid between wheat and rye which was introduced in the 1960’s, and which was initially completely resistant to wheat powdery mildew. We found that the new Triticale mildew (Blumeria graminis forma specialis triticale) originated from hybridization of mildews from wheat and rye. This hybridization occurred at least twice independently already ~50 years ago (i.e. just a few years after the Triticale crop was first introduced.
Additionally, we analyzed a worldwide sample of ~200 wheat powdery mildew genomes and could show that the spread of the disease in historical and modern times tightly follows historical human migration and trade routes. The study also showed that wheat powdery mildew went through multiple hybridization events throughout history: one hybridization happened after wheat was brought to the Americas by Europeans in the 1700s. Here, mildew from Europe hybridized with a distant relative (probably a mildew that grew on barley relatives that were native to America) to form the predominant mildew population found now in America. Additionally, we could show that mildews from Japan are hybrids between mildews of Chinese and American ancestry, and that this hybridization occurred most likely after World War II when wheat from America was introduced to Japanese breeding programs. Our studies identified hybridization and recombination between mildews strains as a major genetic mechanism for crop pathogen adaptation. Additionally, they show agricultural crop breeding and human trade and migration to be essential factors in the spread and emergence of new pathogen variants
Transposable elements
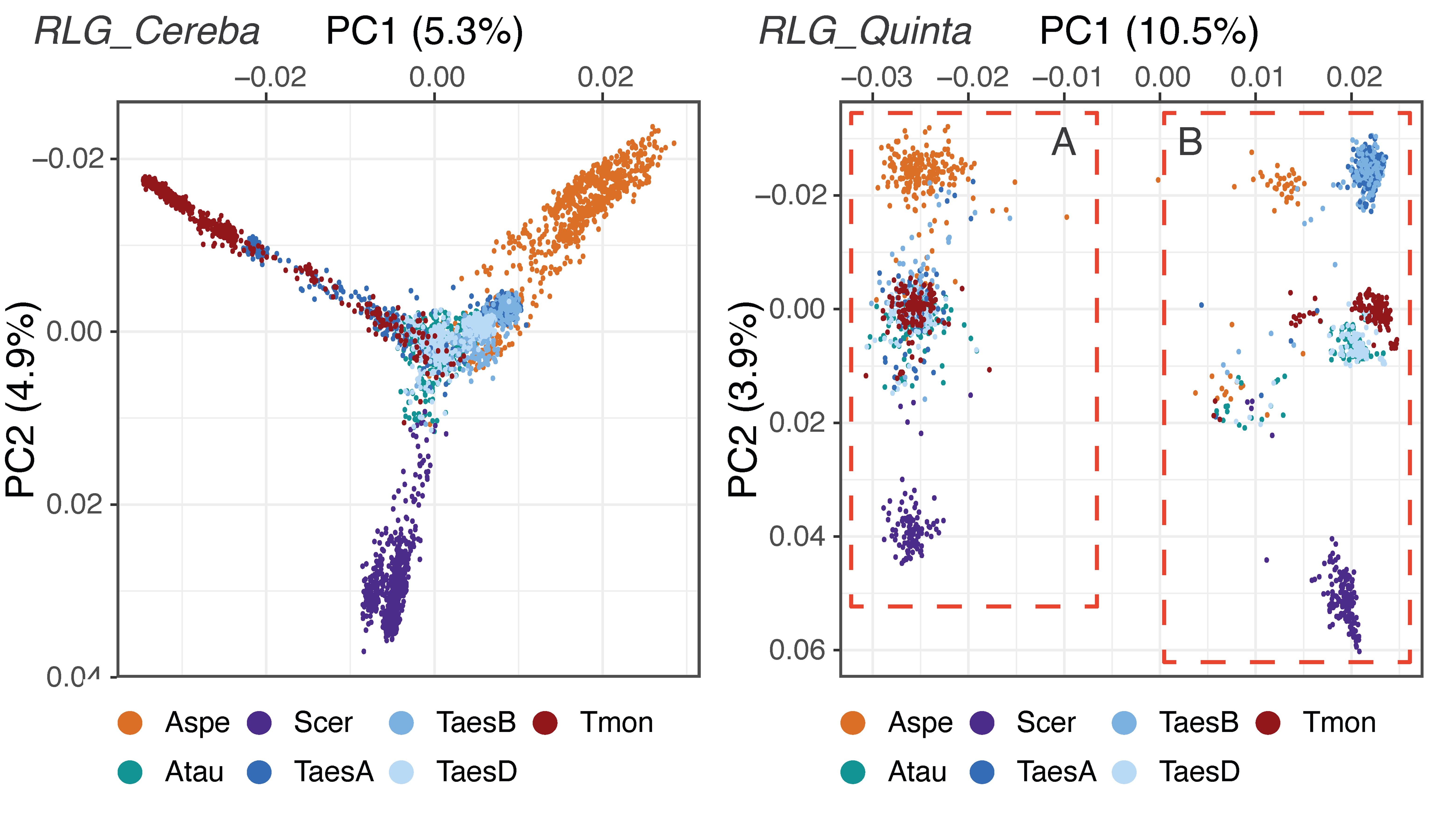
The role of transposable elements (TEs) in genome evolution has long been the over-arching topic of the research in our group. Through comparative analyses, we showed that intergenic regions in plants are rapidly re-shuffled through a balance of TE amplification and removal of sequences through deletions, indicating that genome size is the result of a dynamic equilibrium between TE-driven genome expansion and DNA loss. Interestingly, different types of TEs are found in distinct genomic “niches” leading to an overall compartmentalization of chromosomes. Importantly, we could also show that repair of TE-induced double-strand breaks is a major driver of genetic diversity, for example leading to gene movement or exon shuffling. Additionally, TE insertions and excisions near genes lead to increased mutation rates of gene promoters and coding sequences. Taken together, our findings have identified multiple mechanisms in which TEs contribute to genome evolution.
With more and more complete sequences of large and complex genomes becoming available, entire populations of TEs could be analyzed. Here, individual copies of a TE families are seen as individuals of a population that resides and evolves in its environment, the genome (see example Figure above). Such TE population (TEpop) analyses are highly informative for example for the detection of chromosomal introgressions or for studying gene flow and phylogeny of closely related species.
Interestingly, we also found that particular Gypsy retrotransposons are the main factors that drive the highly dynamic evolution of wheat centromeres. Furthermore, we found evidence that selection acts in multiple ways on the TE fraction, for example in preserving spacing between genes, or providing the genomic environment for the rapid evolution of effector genes in fungal pathogens. TEs also contribute to the evolution of novel functional sequences: we recently found evidence that some miniature inverted-repeat transposable elements (MITEs) were “domesticated” in wheat and produce small RNAs that are involved in host/pathogen interactions. We also found evidence that TEs contribute to the evolution of lineage-specific genes in barley, wheat and rye.
Metagenomics of lichens

In recent years, we have entered the field of metagenomics of lichens. Lichens are highly complex symbiotic systems of several organisms from different domains of life. They are typically a symbiosis formed between lichenized fungi and algae and/or at least one bacteria species. Lichenization has evolved independently multiple times. They are thus important models to study interactions between distantly related organisms. Furthermore, it is not yet clear how many species actually contribute to the symbiosis, and how many are mere opportunists or parasites. In my group, we focus on lichens from the genus Cladonia, among which are the famous reindeer lichens.
Despite numerous studies on microbiome diversity, studies that aim at unraveling entire lichen metagenomes are rare. The main challenge working with metagenome assemblies is that they comprise a complex mixture of sequences from fungi, algae and bacteria. Primary assemblies usually comprise thousands of sequence contigs that then have to be assigned to individual species. Here, taxon-specific characteristics such as GC content, oligonucleotide frequencies and gene content can be used to infer to which species contigs belong.
Considering the limited resources on metagenome sequences in lichens in general and Cladonia in particular, we aim at producing a reference metagenome for C. rangiformis a lichen from Greece. Our goal is to produce chromosome-scale assemblies from the fungus and the green algae. Additionally, we attempt to assemble near-complete genomes of individual bacteria species.